Cystinosis and impaired autophagy/Mitophagy: Cystinosis is a rare autosomal recessive lysosomal storage disorder caused by the mutations in the CTNS gene encoding the lysosomal membrane transporter cystinosin, resulting in the accumulation of toxic metabolite cysteine in the cells. Continuous cystine accumulation eventually leads to multiorgan dysfunction and in the absence of treatment, they usually develop progressive renal failure by the end of the first decade. Treatment with the drug cysteamine depletes the intracellular cystine and if used early in the disease and in high doses it can lower the progression of renal glomerular damage and extra-renal organ injury. We found that cysteamine is ineffective against the proximal tubulopathy, and it delays but cannot avoid the progression of renal injury and failure.
The pathophysiology of cystinosis appears to be more complex than pure cell damage caused by cystine accumulation. We showed that autophagy and mitochondrial pathways have been implicated in the disease pathogenesis and dysregulation of these pathways and their regulating molecules may explain the lack of correlation between genotype and phenotype (disease severity). Currently, we in our lab are exploring for these additional modifier genes and pathways associated with nephropathic cystinosis with a view to also develop new therapies to rescue the renal phenotype in this disease. We are currently evaluating the efficacy of repositioning FDA approved drugs targeting these additional genes. This repurposing strategy will be used to facilitate the fast transfer of “new” therapies against cystinosis to the clinics.
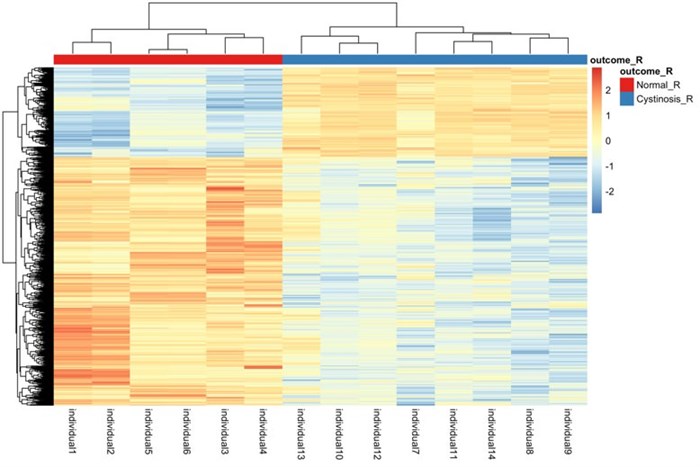
Figure 1
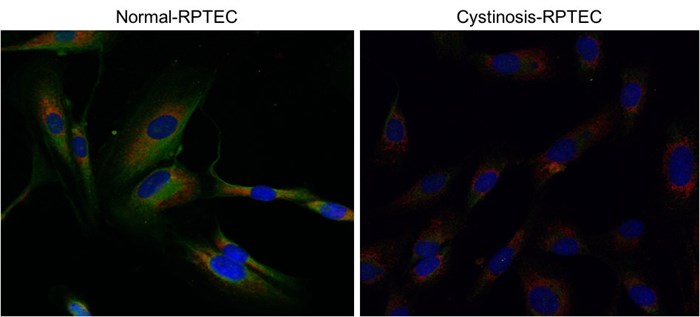
Figure 2
Cystinosis and Male Infertility: In addition to various endocrine organs that can be affected such as kidney, hypergonadotropic hypogonadism has been reported as a frequent finding in male cystinosis patients. As of yet, in contrast to a few female patients who have given birth, no male cystinosis patient is known to have naturally fathered a child. More recently, it was shown that this infertility in male cystinosis patients is due to azoospermia of yet unknown origin.
Although spermatogenesis has shown to be intact at the testicular level in some patients, no male cystinosis patient was reported to have successfully induced conception until recently. This first and the only successful conception ever reported in a male renal transplant infantile nephropathic cystinosis patient is through assisted reproductive technology. To this date, the exact pathophysiology of azoospermia observed in patients with cystinosis is not yet fully understood, which is crucial due to the growing population of cystinosis patients, who were treated with cysteamine is reaching young adulthood. Currently, we are using metabolomics, proteomics, and transcriptomics to obtain a clearer overview of the cellular pathways affected in cystinosis testicular biopsies compared to normal and infertile patients.
At present we are working with Dr Elena Levtchenko, Professor at the Universitair Ziekenhuis Leuven, Leuven and Dr James Smith, Director of the UCSF Male Reproductive Health on various aspects of this project. With Dr Smith, we are developing surveys to reach out to male cystinotic patients and their families.
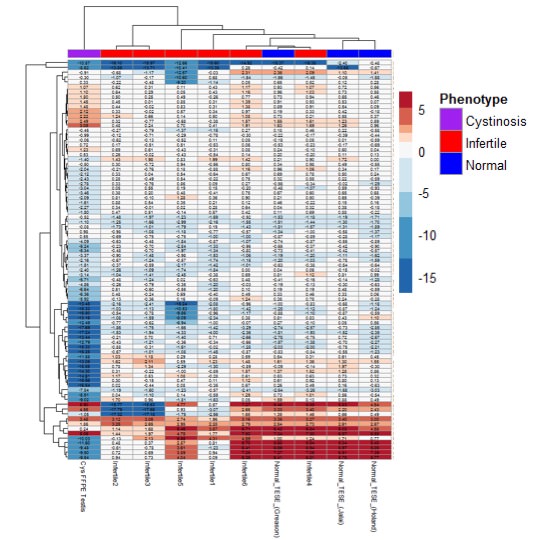
Figure 3